Capítulo II da série de textos sobre o COVID-19
SARS-CoV-2: Virologia
Autores:
Cláudio Silva: Unidade de Doenças Emergentes, Serviço de Doenças Infeciosas, Centro Hospitalar Universitário de São João, Porto
Margarida Tavares: Unidade de Doenças Emergentes, Serviço de Doenças Infeciosas, Centro Hospitalar Universitário de São João, Porto e EPIUnit, Instituto de Saúde Pública da Universidade do Porto
1. Um novo coronavírus humano
Os coronavírus são vírus de ARN simples de sentido positivo amplamente distribuídos no ser humano e noutros mamíferos. O tamanho do genoma dos coronavírus é o maior dentro dos vírus de ARN. Por outro lado, as taxas de mutação durante as fases iniciais de replicação e os fenómenos de recombinação dos vírus de ARN é muito maior do que as dos vírus de ADN, resultando na produção de uma maior diversidade de proteínas estruturais com capacidade de adaptação a diferentes ambientes. A proximidade do ser humano com o reservatório animal associado à alta prevalência e distribuição dos coranavírus, à sua diversidade genética e aos fenómenos de recombinação frequentes dos seus genomas, justificam o aparecimento periódico de novos coronavírus capazes de ultrapassar a barreira entre espécies e causar infeções no ser humano. Foi mais uma vez o que aconteceu com a identificação de um novo coronavírus como causa de um surto de pneumonia com início no final do ano de 2019 na cidade de Wuhan, o 2019-nCoV, nome atribuído de forma temporária no dia 12 de Janeiro de 2020 pela OMS.
O novo vírus identificado neste surto pertence à subfamília Coronovirinae dentro da família Coronaviridae da ordem Nidovirales. Nesta sub-família incluem-se 4 géneros de coronavírus: Alphacoronavirus, Betacoronavirus, Gammacoronavirus e Deltacoronavirus. Destes, sabe-se que apenas os primeiros dois géneros são capazes de infetar o ser humano. Quatro coronavírus são endémicos globalmente (HCoV-229E, HCoV-NL63, HCoV-OC43 e HCoV-HKU1) e contribuem para 10-30% dos casos de infeções das vias respiratórias superiores. Duas outras espécies, SARS-CoV e MERS-CoV, foram reconhecidos como vírus emergentes com origem em reservatórios animais, e causa de surtos de infeções respiratórias em humanos em 2003 e 2012, respetivamente. O tropismo que as proteínas de superfície (S) destas últimas espécies apresentam para recetores presentes no trato respiratório inferior justifica a apresentação na forma de pneumonia vírica potencialmente grave que provocam, incluindo a síndrome de dificuldade respiratória aguda.
1.1 Nova nomenclatura para o 2019-nCoV: o SARS-CoV-2 será uma espécie de vírus distinta da de outros coronavírus isolados previamente?
É difícil definir um vírus de ARN como pertencente a uma espécie nova (ou distinta): devido às suas altas taxas de mutação e fenómenos de recombinação à medida que a replicação e seleção viral prossegue, é natural que surjam variantes de determinadas sequências definidas (haplótipos), também conhecidos como quasispécies. A sua sequência genómica está em contínua evolução e pode variar entre diferentes hospedeiros infetados ou mesmo ao longo de um surto. Assim, do ponto de vista da virologia, se usarmos o critério “novo” para designar um coronavírus cuja semelhança genómica é incompleta face a outros coronavírus isolados previamente, qualificaríamos qualquer vírus de ARN após sequenciação total do seu genoma como tal. Para contornar este problema, os virologistas olham para dois vírus com genomas não idênticos, embora similares, como variantes do mesmo vírus, colocando a questão: “quão grande deve ser a diferença de um vírus para classificá-lo como pertencente a uma espécie nova ou distinta?”. No que diz respeito ao vírus identificado neste surto, esta questão é respondida pelo Grupo de Estudo dos Coronavírus do Comité Internacional de Taxonomia dos Vírus (ICTV) através da avaliação do grau de proximidade do vírus candidato em relação a coronavírus previamente isolados em humanos e em relação a grupos monofiléticos de coronavírus (clades) que podem ou não incluir vírus de diferentes hospedeiros.
A classificação dos vírus pertencentes à ordem dos Nidovirales pelo ICTV atualmente baseia-se na análise comparativa de sequências dos domínios de proteínas envolvidas na replicação: 3CLpro, NiRAN, RdRp, ZBD e HEL1, os únicos domínios codificados pelas ORF1a/1b conservados em todos os vírus pertencentes a esta ordem. Desde 2011 que a classificação dos coronavírus e outros nidovírus tem sido ajudada pelo software DEmARC (DivErsity pArtitioning by hieRarchical Clustering), que delimita grupos (clusters) monofiléticos de vírus de acordo com distâncias patrísticas.
A designação 2019-nCoV inicialmente atribuída ao novo coronavírus identificado na China no final do ano de 2019 foi formalmente alterada para SARS-CoV-2 pelo ICTV no dia 11 de fevereiro de 2020. O SARS-CoV-2 (nome pelo qual será referido daqui em diante) agrupa-se com SARS-CoVs nas árvores de espécies de coronavírus relacionados com o SARS (Severe acute respiratory syndrome-related coronavirus) e nas árvores de vírus pertencentes ao género Betacoronavirus (figura 1). A demarcação de espécies dentro da família Coronaviridae é imposta por vírus cuja distância patrística cruze o limite de demarcação inter-espécies (figura 2). Nenhum vírus pertencente à espécie de coronavírus relacionados com SARS viola este limite de demarcação como demonstrado numa análise de diferenças patrísticas máximas intra-espécie de 2505 vírus pertencentes a todas as 49 espécies de coronavírus conhecidas (figura 2) e numa análise de diferenças genéticas emparelhadas de 256 vírus pertencentes à espécie de coronavírus relacionados com SARS (figura 3), facilitando a atribuição da designação SARS-CoV-2 a esta espécie. Apesar de não cruzar o limite inter-espécies baseado na distância patrística, o não isolamento inicial do SARS-CoV-2 utilizando técnicas de PCR específicas para o SARS-CoV juntamente com a sua distância filogenética em relação ao último levou a que muitos investigadores – “erradamente” – o considerassem como um vírus pertencente a uma espécie distinta (“nova”) dentro dos coronavírus.
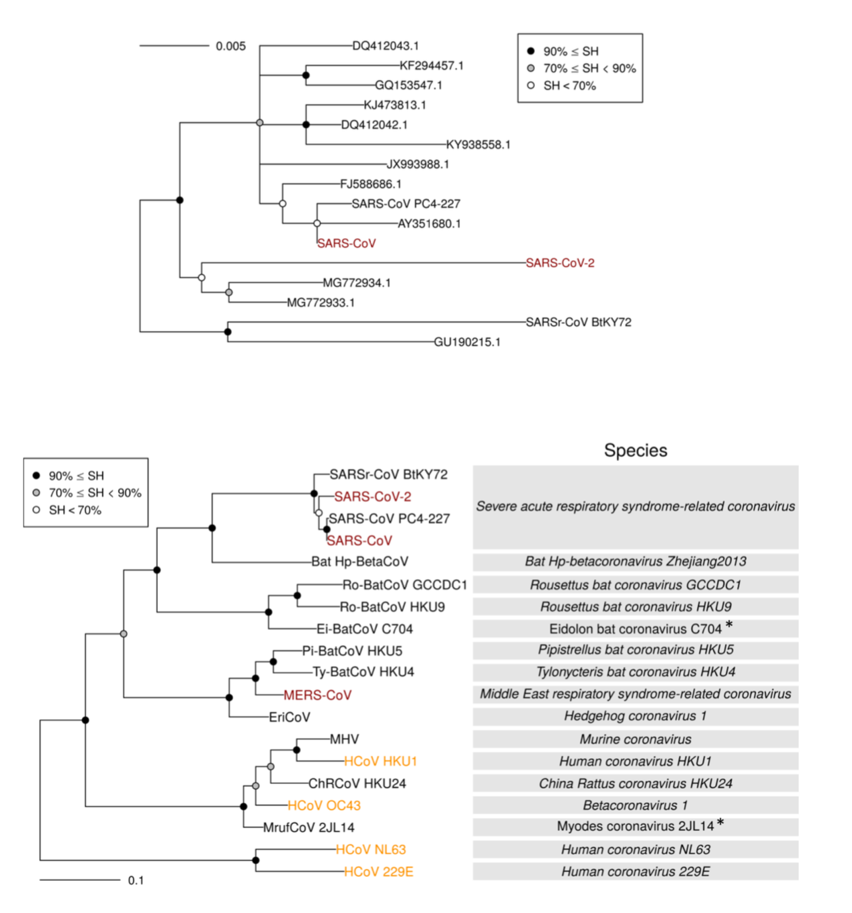
Figura 1.O SARS-CoV-2 agrupa-se com SARS-CoVs nas árvores de espécies de coronavírus relacionados com o SARS (Severe acute respiratory syndrome-related coronavirus) e nas árvores de vírus pertencentes ao género Betacoronavirus.
A demarcação de espécies dentro da família Coronaviridae é imposta por vírus cuja distância patrística cruze o limite de demarcação inter-espécies (figura 2). Nenhum vírus pertencente à espécie de coronavírus relacionados com SARS viola este limite de demarcação como demonstrado numa análise de diferenças patrísticas máximas intra-espécie de 2505 vírus pertencentes a todas as 49 espécies de coronavírus conhecidas (figura 2) e numa análise de diferenças genéticas emparelhadas de 256 vírus pertencentes à espécie de coronavírus relacionados com SARS (figura 3), facilitando a atribuição da designação SARS-CoV-2 a esta espécie. Apesar de não cruzar o limite inter-espécies baseado na distância patrística, o não isolamento inicial do SARSCoV-2 utilizando técnicas de PCR específicas para o SARS-CoV juntamente com a sua distância filogenética em relação ao último levou a que muitos investigadores — “erradamente” — o considerassem como um vírus pertencente a uma espécie distinta (“nova”) dentro dos coronavírus.
Assim, em termos de classificação taxonómica, o SARS-CoV-2 é considerado como mais um vírus dentro da espécie de vírus relacionados com o SARS. Todos os vírus pertencentes a esta espécie têm nomes derivados do SARS-CoV apesar de apenas as estirpes isoladas em humanos colhidas durante 2002-2003 terem causado a clínica de SARS (síndrome respiratória aguda grave) nos indivíduos infetados. A referência ao SARS em todos os nomes dos vírus pertencentes a esta espécie tem em consideração o seu agrupamento filogenético com a dos vírus isolados nos doentes com SARS (exemplo: SARS-CoV-Urbani) e não a sua ligação com determinada síndrome clínica.
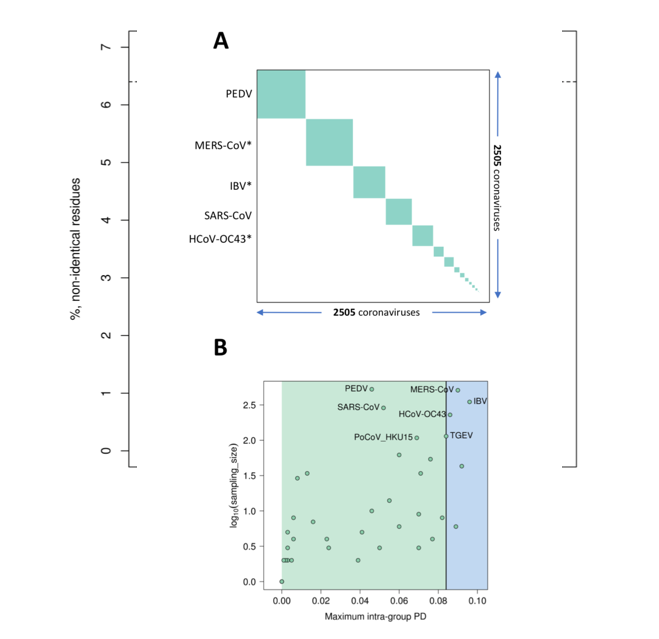
Figura 2. A demarcação de espécies dentro da família Coronaviridae é imposta por vírus cuja distância patrística cruze o limite de demarcação inter-espécies (A). Nenhum vírus pertencente à espécie de coronavírus relacionados com SARS viola o limite de demarcação inter-espécie como demonstrado numa análise de diferenças patrísticas máximas intra-espécie de 2505 vírus pertencentes a todas as 49 espécies de coronavírus conhecidas (B).

Figura 3. Nenhum vírus pertencente à espécie de coronavírus relacionados com SARS viola o limite de demarcação inter-espécie numa análise de diferenças genéticas emparelhadas de 256 vírus pertencentes à espécie de coronavírus relacionados com SARS.
2. Origem animal do SARS-CoV-2
A sequenciação genómica do SARS-CoV-2 a partir de amostras obtidas de 9 doentes infetados em Wuhan revela uma semelhança global de 87.99% e 87.23% com a de outros dois coronavírus do tipo SARS previamente isolados em morcegos em 2018 em Zhoushan, na China oriental (bat-SL-CoVZC45 e bat-SL-CoVZXC21, respetivamente). De notar que as 9 estirpes de SARS-CoV-2 isoladas são geneticamente menos semelhantes ao SARS-CoV (cerca de 79%) e MERS-CoV (cerca de 50%). A análise filogenética (figura 4) revelou que o SARS-CoV-2 (na figura denominado como 2019-nCoV) pertence assim ao grupo monofilético (clade) 2 do subgénero Sarbecovirus dentro do género Betacoronavirus, com um ramo relativamente longo separando-o dos seus “parentes” mais próximos (bat-SL-CoVZC45 e bat-SL-CoVZXC21), assente no facto da semelhança global dos seus genomas ser < 90%.
Dada a sua semelhança com estes coronavírus previamente isolados em morcegos, crê-se que estes constituem o reservatório natural deste vírus. No entanto, e apesar da sua importância como reservatório natural, acredita-se que outro animal vendido no mercado de Huanan atuou como hospedeiro intermediário. De facto, a origem deste surto remonta aos finais de dezembro de 2019, altura do ano em que a maioria das espécies de morcegos hibernam em Wuhan. Por outro lado, este novo coronavírus pertence a outra clade, sugerindo que os morcegos não constituem os seus ancestrais diretos. A 7 de fevereiro de 2020, numa conferência de imprensa, a Universidade de Agricultura do Sul da China, em Guangzhou, revelou que 2 dos seus investigadores, Shen Youngyi e Xiao Lihua, identificaram o pangolim como potencial hospedeiro intermediário do SARS-CoV-2 com base na semelhança de 99% entre as sequências genómicas dos coronavírus isolados nestes animais e em humanos infetados durante o surto atual. De referir que, em 2019, tantos os coronavírus como o vírus Sendai (da família Paramyxoviridae) foram identificados como causadores de mortalidade em pangolins malaios.
3. Ligação do SARS-CoV-2 a células humanas
A proteína S do envelope dos coronavírus é dividida nos domínios S1, responsável pela ligação aos recetores de células humanas e S2, responsável pela fusão subsequente das suas membranas. O domínio S2 do SARS-CoV-2 demonstra uma semelhança de cerca de 93% com o dos bat-SL-CoVZC45 e bat-SL-CoVZXC21, muito maior que a do domínio S1, que apenas apresenta uma semelhança de 68%. Uma análise das variações aminoacídicas da proteína S nos coronavírus pertencentes ao subgénero Sarbecovirus revela que tanto o SARS-CoV-2 como o SARS-CoV apresentam perto de 50 aminoácidos conservados entre si ao nível do domínio S1 (enquanto que a maioria dos coronavírus provenientes dos morcegos apresentam mutações importantes neste domínio). É assim possível concluir que o domínio de ligação aos recetores de células humanas do SARS-CoV-2é mais parecido com o do SARS-CoV, sugerindo que este, à semelhança do último, utilize a enzima conversora da angiotensina 2 – ACE2 (presente nas células epiteliais dos alvéolos pulmonares) como recetor humano.

Figura 4. Análise filogenética dos genomas completos do 2019-nCoV e outros Betacoronavirus
As referências do artigo encontram-se disponíveis no PDF do documento do “Capítulo II: SARS-CoV-2: Virologia_27-02-2020″.